Allgemeine Theorie der Phasentransfer-Katalyse
Einleitung
Die Phasentransfer-Katalyse (PTC) ist eine relativ neue präparative Methode der Chemie. Die
Grundlagen wurden in den späten 60'er Jahren von drei unabhängigen Arbeitsgruppen
(M. Makosza, C. M. Starks und A. Brändström) gelegt.
Phasentransfer-Katalysatoren werden für Reaktionen zwischen Spezies in unterschiedlichen Phasen
eingesetzt. Gewöhnlich findet die Reaktion zwischen einem Salz und in organischen Lösungsmitteln
gelösten Substanzen statt. Dabei kann das Salz sowohl in wäriger Lösung als auch ungelöst als
Feststoff vorliegen. Ohne Katalysator sind solche Reaktionen in der Regel langsam, treten überhaupt
nicht auf oder ergeben ein anderes Produktspektrum als mit Katalysator.
Als Katalysatoren kommen Onium-Salze oder komplexierende Substanzen, die Alkalimetall-Ionen
maskieren und damit lösen können, zum Einsatz. Die Funktion des Katalysators besteht meistens
darin, Anionen des Salzes als Ionenpaar in die organische Phase zu transportieren. In aprotischen
Lösungsmitteln liegen diese Ionenpaare wenig solvatisiert vor und sind sehr reaktiv.
Somit bietet die Phasentransfer-Katalyse folgende Vorteile gegenüber anderen
präparativen Methoden:
- Einsparungen an teuren, wasserfreien, aprotischen Lösungsmitteln
- verkürzte Reaktionszeiten und / oder
- niedrigere Reaktionstemperaturen
- vereinfachte Aufarbeitung
- Verwendung von wärigen Alkalimetallhydroxiden an Stelle von Alkalimetallalkoxiden,
Natriumamid, Natriumhydrid oder metallischem Natrium.
- Auffinden von Reaktionen, die anders nicht durchführbar sind
- Verschiebung der Selektivität
- Änderung von Produktverhältnissen (z.B. O-/C- Alkylierung)
- erhöhte Ausbeuten durch Unterdrückung von Nebenreaktionen
Mechanismus der Phasentransfer-Katalyse
Reaktionen unter Phasentransfer-Katalyse können unter sauren, neutralen oder basischen Bedingungen
durchgeführt werden.
Unter neutralen Bedingungen
Die PTC-Reaktion für nucleophile Substitutionen wird in
Abbildung 1 aufgezeigt.
Es wird ein von Starks
vorgeschlagener Mechanismus angenommen. Dabei
wird vorausgesetzt, da Y- lipophiler als X- ist. Das Katalysator-Kation
Q+ wandert mit dem Anion Y- von der wärigen in die organische Phase. Dort reagiert
das entstandene, schwach solvatisierte Ionenpaar schnell mit RX.
Q+ kehrt mit X- in die
wärige Phase zurück, wo Q+ ein neues Y- anlagern kann.
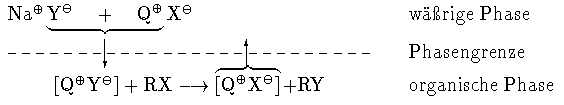
Abbildung 1: Ablauf der PTC-Reaktion bei nucleophilen Substitutionen (Ionenpaare in eckigen
Klammern).
Werden sehr lipophile Katalysator-Kationen verwendet, so liegen keine merklichen
Konzentrationen von Q+ im wärigen Medium vor und es werden nur die Anionen
über die Phasengrenze hinweg ausgetauscht. Der Mechanismus vereinfacht sich zu dem in
Abbildung 2 gezeigten.
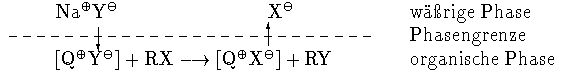
Abbildung 2: Ablauf der PTC-Reaktion bei nucleophilen Substitutionen und lipophilem Katalysator-Kation.
In Gegenwart von Alkalimetallhydroxiden
Bei Reaktionen in Gegenwart von starken Basen ( z.B. bei C-, O- und
N- Alkylierungen) ist der Mechanismus nicht so eindeutig, er hängt vom
pKa Wert der C-H, N-H oder O-H Säure ab. Relativ starke Säuren, wie Acetylaceton
(pKa~9)lösen sich in Natronlauge.
Der Phasentransfer-Katalysator dient dazu, das Anion als Ionenpaar in die organische Phase
zurückzuextrahieren, wo dann die C- oder O- Alkylierung stattfinden kann. Es gilt
wieder der Mechanismus aus Abbildung 2.
Für PTC-Alkylierungen schwacher Säuren wurde folgender Mechanismus postuliert:
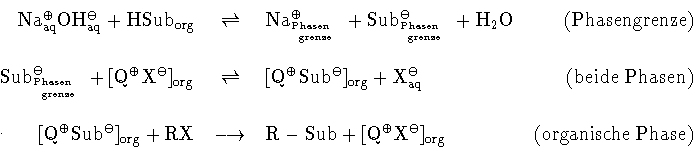
Die Deprotonierung findet dabei an der Phasengranze statt. Das Katalysator-Kation
erleichtert durch seine positive Ladung die Deprotonierung des Substrates. Auerdem unterstützt es
die Ablösung des Substrat-Anions von der Phasengrenze.
Der Einflu des Lösungsmittels
Die Bildung von Ionenpaaren und ihre physikalischen und chemischen Eigenschaften werden
stark von den Wechselwirkungen mit dem Lösungsmittel bestimmt:
- Polare protische Lösungsmittel
- (z.B. Alkohole) solvatisieren sowohl Anionen als auch Kationen.
Jedoch bleiben groe quartäre Ammoniumsalze ungelöst. Wegen der Abschirmung durch die
Solvathülle verhalten sich viele Anionen wenig reaktiv.
- Polare aprotische Lösungsmittel
- (z.B. DMSO, DMF) solvatisieren Kationen gut, Anionen aber
nur schwach. Daher liegen Salze stark dissoziiert vor und sind somit sehr reaktiv.
- Wenig polare aprotische Lösungsmittel
- lösen anorganische Salze kaum.
Quartäre Oniumsalze und maskierte Alkalimetallsalze sind aber oft gut löslich. Da die
Wechselwirkungen zwischen Lösungsmittel und Ionen gering sind, liegen diese fast ausschlielich als
Ionenpaare vor und sind sehr reaktiv.
Ein für die Phasentransfer-Katalyse geeignetes Lösungsmittel sollte nicht mit Wasser mischbar
und aprotisch sein. Andererseits sollte es in der Lage sein, eine genügend groe Menge Ionenpaare zu
extrahieren.
Als besonders geeignete Lösungsmittel haben sich Dichlormethan,
Chloroform und 1,2-Dichlorethan herausgestellt, die oft auch kleine organische Kationen (z.B.
Tetramethylammonium) lösen können.
Fest/flüssig PTC wird oft in Toluol, Chlorbenzol oder in einem von den
genannten halogenierten Lösungsmitteln durchgeführt. Reaktionen ohne
zusätzliches Lösungsmittel sind ebenfalls möglich.
Einflu des Onium-Kations auf die Extraktion
Onium-Kationen unterscheiden sich durch ihre Lipophilie und damit durch ihre Extraktionskonstante
EQX für das Extraktionsgleichgewicht zwischen organischer und wäriger Phase.
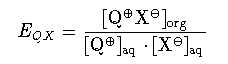 Eine Erhöhung der Zahl der Kohlenstoffatome, die das Zentralatom im Onium-Kation umgeben, führt zu
einer Steigerung der Lipophilie. Ein Anstieg von log EQX um etwa 0.54 bis 0.61 pro
Kohlenstoffatom bei Ammoniumionen wurde in verschiedenen Untersuchungen gefunden.
Die Zahl der Kohlenstoffatome ist aber nicht der einzige Faktor, der die Lipophilie bestimmt. So
sind z.B. Benzylgruppen viel weniger lipophil als 1-Heptylgruppen und liegen, was ihren Beitrag
zur Extraktionskonstante angeht, zwischen Butyl- und Propylgruppen.
Deshalb liegt Benzyltriethylammoniumchlorid (TEBA-Cl) hauptsächlich in der wärigen Phase vor.
Bei Phosphonium- und Arsoniumionen liegen ähnliche Trends vor. Folgende lipophile Reihe wurde
aufgestellt:
Eine Erhöhung der Zahl der Kohlenstoffatome, die das Zentralatom im Onium-Kation umgeben, führt zu
einer Steigerung der Lipophilie. Ein Anstieg von log EQX um etwa 0.54 bis 0.61 pro
Kohlenstoffatom bei Ammoniumionen wurde in verschiedenen Untersuchungen gefunden.
Die Zahl der Kohlenstoffatome ist aber nicht der einzige Faktor, der die Lipophilie bestimmt. So
sind z.B. Benzylgruppen viel weniger lipophil als 1-Heptylgruppen und liegen, was ihren Beitrag
zur Extraktionskonstante angeht, zwischen Butyl- und Propylgruppen.
Deshalb liegt Benzyltriethylammoniumchlorid (TEBA-Cl) hauptsächlich in der wärigen Phase vor.
Bei Phosphonium- und Arsoniumionen liegen ähnliche Trends vor. Folgende lipophile Reihe wurde
aufgestellt:
NHex4+ > NPent4+ >=
AsPh4+ > N(iso-C5H11)4+ >
PBu4+ > NBu4+ .
Kronenether
Für die Phasentransfer-Katalyse interessant sind Komplexe aus Kronenethern und Natrium- oder
Kaliumionen. Die stabilsten Kaliumkomplexe bilden sich mit Kronenethern, die einen 18 Atome groen
Ring besitzen wie 18-Krone-6, Dibenzo-18-krone-6 und Dicyclohexyl-18-krone-6. Das kleinere
Natriumion bindet sich ebenfalls stärker an die 18 Atome groen Ringe als an die kleineren
vergleichbaren Kronenether wie 15-Krone-5, allerdings weniger stark als das
Kaliumion.
Kronenether können deshalb als Phasentransfer-Katalysatoren wirken, weil sie
Alkalimetallionen "organisch maskieren" und sie zusammen mit ihrem Anion in unpolare
organische Lösungsmittel extrahieren können. Der anionische Teil dieses
Ionenpaares ist aktiviert. Kronenether schirmen die positive Ladung des Kations ab und
fördern somit in polaren aprotischen Lösungsmitteln die Dissoziation des
Ionenpaares. In weniger polaren Lösungsmitteln begünstigen sie die Bildung
von lockeren, durch Lösungsmittelmoleküle getrennten Ionenpaaren mit
höherer Reaktivität. Sogar in protischen Lösungsmitteln können
Kronenether durch änderung von Struktur und Zusammensetzung der
Solvathülle zu einer Aktivierung führen.
Einflu des Anions
Der Einflu des Anions auf die Extraktion wurde in mehreren Arbeiten unter unterschiedlichen
Bedingungen beschrieben und führte zu folgender lipophiler Reihe:
Picrat >> MnO4- > ClO4- >
SCN- > I- > ( ClO3-, Toluolsulfonat )
> NO3- > Br- > ( CN-, BrO3-,
Benzoat ) > ( NO2-, Cl- )> HSO4-
> (HCO3-, Acetat ) > (F-, OH-) > SO4-
> CO3- > PO4-
Bei PTC-Reaktionen findet eine konkurrierende Extraktion zwischen Katalysatoranion und dem
Substratanion statt.
Sehr lipophile Anionen wie Iodid und Perchlorat werden bevorzugt extrahiert, was dazu
führen kann, da das Kation als Ionenpaar zusammen mit dem lipophilen Anion
extrahiert wird und nicht mehr als Katalysator wirken kann. Ein sehr lipophiles Anion
kann also den Katalysator "vergiften", während ein wenig lipophiles Anion die
Extraktion des Substratanions begünstigt.
Einflu der Katalysatormenge
üblich sind Katalysatormengen von 0.5-10 mol%. Eine typische PTC-Reaktion zeigt
eine lineare Abhängigkeit der Geschwindigkeit von der Katalysatormenge.
Phasentransfer-Katalysatoren können aber besonders in wenig polaren
Lösungsmitteln auch Micellen bilden, was zu einem starken Anstieg der
Reaktionsgeschwindigkeit in der Nähe der "kritischen Micell-Konzentration"
führt.
Rührgeschwindigkeit
Um reproduzierbare kinetische Ergebnisse zu erhalten, mu die Rührgeschwindigkeit
oberhalb eines Minimalwertes liegen, der sicherstellt, da Konzentrationsgradienten auf
beiden Seiten der Phasengrenze verhindert werden. Unter Laborbedingungen sind mit einem
Magnetrührer bei verdünnten Lösungen Geschwindigkeiten von 200/min und
bei konzentrierteren Lösungen 750/min ausreichend .
Literatur
- Starks, C. M., J. Am. Chem. Soc. 93, 195 (1971).
- Dehmlow, E. V., Dehmlow, S. S., Phase Transfer Catalysis, Verlag Chemie, 3. erweiterte Auflage, 1993.
- Starks, C. M., Liotta, C., Phase Transfer Catalysis, Principles and Techniques, Academis Press, London and New York
(1978).
- Weber, W. P., Gokel, G. W. "Phase Transfer Catalysis in Organic Synthesis" in:
Reactivity and Structure, K. Haffner et. al.,Eds., Springer Verlag, Berlin, Heidelberg, New York (1977).
- W. P., Gokel, G. W., J. Chem. Educ. 55, 350, 429 (1978).
- Dehmlow, E. V., Angew. Chem. 86, 187 (1974).
- Dehmlow, E. V., Angew. Chem. 89, 521 (1977).
- Dockx, J., Synthesis, 441 (1973).
- Makosza, M., Pure Appl. Chem. 43, 439 (1975).
- Brändström, A., Preperative Ion Pair Extraction, an Intodruction to Theory and Practice,
Apotekasocieteten/Hässle Läkemel, Stockholm (1974).
- Brändström, A., "Principles of Phase Transfer Catalysis by Quaternary Ammonium
Salts", in: Adv. Phys. Org. Chem. 15, 267 (1977).
- Starks, C. M., Owens, R. M., J. Am. Chem. Soc. 95, 3613 (1973).
- Brändström, A., Adv. Phys. Org. Chem. }15, 267 (1977).
- le Noble, W. J., in: Highlights of Organic Chemistry, Marcell Dekker Inc., New York (1974), S. 821 ff.
- Brändström, A., Junggren, U., Acta Chem. Scand. 25, 1469 (1971).
- Gustavii, K., Acta Pharm. Suec. 4, 233 (1967).
- Gustavii, K., Schill, G., Acta Pharm. Suec. 3, 241 (1966).
- Antoine, J. P., de Aguirre, I., Janssens, F.,Thyrion, F., Bull. Soc. Chim. Fr. , 207 (1980).
- Herriot, A.W., Picker, D., J. Am. Chem. Soc. 97, 2345 (1975).
1994 by Robert Klauck 